Here is an post-mortem analysis / "how to" for the Final. The questions are split by the sections. At the start of each section are a few suggestions of what to look for or how to tackle the question type.
RELATIVE PROPERTIES:
Identify the controlling feature, which is not
always as obvious as it may appear. Look for two pairs of similar systems to compare
that have minimal differences in structure. If a compound is named, draw it out.
If a reaction is involved, identify the type of reaction and then what the controlling
factors are.
Qu1:
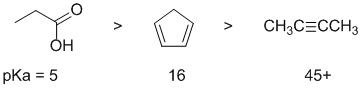
Acidity...Remember the lower the pKa the stronger the acid, and think of the simple acid equation HA <=> H+ A- then look for factors that stabilise A-.....Here we have one O-H and two C-H systems. We have a carboxylic acid, an internal alkyne and a cyclic diene. In a carboxylic acid, the loss of the H+ generates a -ve O where there is also resonance stabilisation to give another -ve O (i.e. delocalisation of the -ve charge across two electronegative O atoms), pKa = 5. For the internal alkyne, the C-H bond is an sp3 C-H bond and there is minimal stabilisation of the -ve charge (note terminal alkynes are a different story). For cyclopentadiene, the conjugate base is aromatic and the resonance stabilisation by the aromatic system is significant, pKa = 16. If you know your pKa's then this is easy : carboxylic acid about 5, internal alkyne about 45(+) and cyclopentadiene about 16. So for strongest acid to weakest acid:
Qu2:
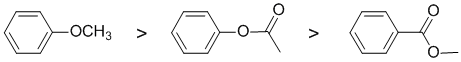
The reaction is electrophilic aromatic substitution, nitration and we need to look at the substituent effects on the aromatic ring. The substituents are a methoxy group, and two esters that differ in the orientation of the attachment. The simple alkoxy group (-OR) is a strong electron donating group via resonance donation of the O lone pair - it's a strongly activating group. If we compare the two ester groups, one is attached through the alkoxy O and therefore is a moderate electron donating group via resonance donation of the O lone pair (note that there are competing effects for the interaction of the lone pair towards the arene and the carbonyl groups) - it's a moderately activating group and the other is attached via the carbonyl group which is deactivating due to inductive and resonance effects.. So the reactivity:
Qu3:
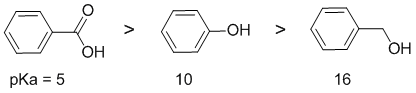
Solubility in aqueous media depends on polarity. Sodium bicarbonate is a weak base and will deprotonate acids making the more polar conjugate base which is more water soluble. Therefore the more acidic these similar structures are, the more soluble they will be in this solution. Here we have a carboxylic acid, a phenol and an alcohol. Acidity...Remember the lower the pKa the stronger the acid, and think of the simple acid equation HA <=> H+ A- then look for factors that stabilise A-. Here it is the possibility and the nature of the resonance delocalisation that influences the stability of the conjugate base.
Qu4:
The reactivity of C=C pi bonds towards sulfuric acid so we are looking at electrophilic addition to an alkene which will be controlled by the stability of the intermediate carbocation. A closer look at the alkene structures shows that each of the alkenes has a different substituent attached: a chlorine, an alkyl group -R, and an phenyl group -Ph. Each alkene will react with the acid to give a carbocation and the substituent will affect the carbocation stability.... an electron donor will stabilise a carbocation, an electron withdrawer will destabilise a carbocation. Looking at the substituents: a chlorine is electron withdrawing, an alkyl group, -R, is a weak electron donor, and a phenyl group, -R, is a weak electron donor but gives us a resonance stablised carbocation (a benzylic system) where the +ve charge can be delocalised to 3 other C atoms.
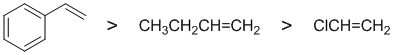
Qu5:
Acidity...Remember the lower the pKa the stronger the acid, and think of the simple acid equation HA <=> H+ A- then look for factors that stabilise A-.... Here we are looking at carbonyl systems and the formation of enolates by the removal of H from the C atom adjacent to the C=O (known as the alpha position) since this allows for resonance stabilisation of the -ve charge. In a simple ketone, such as propanone, the pKa is about 20. . In a 1,3-dicarbonyl, it is the position between the two carbonyls that is the most acidic because the -ve charge can be delocalised to each of the carbonyl groups thus spreading the charge out further (this 1,3-diketone has a pKa = 9). Finally, benzaldehyde which is not just a simple alkyl aldehyde (which would have a pKa = 17) because benzaldehyde does not have alpha-H and is therefore less acidic than the other carbonyl systems.
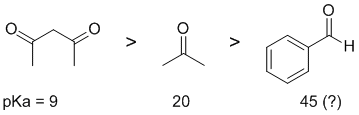
Qu6:
Resonance energy indicates the stability of the conjugated system compare to a non-conjugated system with the same number and type of pi bonds. Aromatic systems have high resonance stablisation and therefore high resonance energies. Conjugated systems also have resonance stabilisation but not to the same extent as aromatics. Of the systems asked, first we have styrene (phenylethene) which contains a benzene ring (perhaps our "model" aromatic compound, has 3 C=C with resonance energy = 36 kcal/mol) which is conjugated with an alkene (and hence further stabilised). Cycloopenta-1,3-diene is not aromatic but it is a conjugated diene (resonance energy about 3 kcal/mol). Cyclohexa-1,4-diene which is not aromatic and not conjugated diene so no resonance stabilisation. Hence:
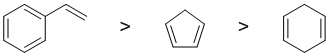
Qu7:
Oxidation states...The easiest way to reach the answer is based on the knowledge that aldehydes can be reduced to primary alcohols, aldehydes can be oxidised to carboxylic acids and that esters are derivatives of carboxylic acids (and at the same oxidation state). More formally, we count the bonds attached to the atom being considered. A bond to a more electronegative atoms counts -1, a bond to the same type of atom counts 0 and a bond to a less electronegative atom (e.g. H) counts +1. Total the count and then consider the formal charge on the central atom since the oxidation state for the central atom plus the groups attached must equal the atoms formal charge. For the aldehyde, the C is attached to 2 x O (count - 1 each, total -2) and H (count +1) therefore total = -1 and therefore the oxidation state C = +1. In the primary alcohol, the C is attached to 1 x O (count -1), 1 x C (count 0) and 2 x H (count +2) therefore total = +1 and therefore the oxidation state C = -1. In the ester the C is attached to 1 x C (count 0) and 3 x O (count -3) therefore total = -3 and therefore the oxidation state C = +3.
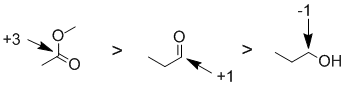
Qu8:
Acidity...Remember the lower the pKa the stronger the acid, and think of the simple acid equation HA <=> H+ A- then look for factors that stabilise A-.....Here we have a series of phenols and need to look at the substituent effects on the aromatic ring. Electron withdrawing groups will help stabilise the conjugate base (the substituted phenoxide) and therefore make the parent phenol more acidic. The bromine is a halogen which is weakly electron withdrawing (electronegativity), the nitro group is strongly electron withdrawing (inductive and resonance) and the alkoxy group (-OR) is a strong electron donating group via resonance donation of the O lone pair. So the acidity is:

Qu9:
The reactivity of carbonyl groups of an aldehyde, an amide and an ester towards a Grignard reagent. The electronic factors of each of the substituents on each of the carbonyl groups need to be considered. In the aldehyde we have a neutral H atom (i.e. no electronic effect). In the ester, the -OR group, the methoxy group is a strong electron donor. In the amide, the NR2 group, is an even stronger electron donating group. Electron donors on the carbonyl make the carbon less electrophilic and less reactive. Hence in terms of reactivity:
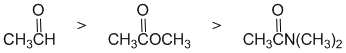
Qu10:
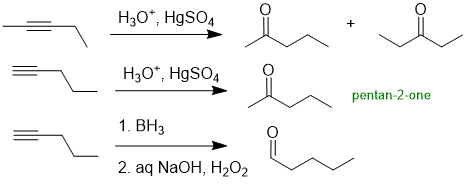
The reaction is alkyne hydration. and its regioselectivity. Pentan-2-one is a methyl ketone. Aq. hydration of a simple terminal alkyne typically gives methyl ketones as the major product ("Markovnikov"). while simple unsymmetrical internal alkynes give mixtures of products. In contrast, hydroboration / oxidation of simple terminal alkyne typically gives aldehydes as the major product ("anti-Markovnikov").
STRUCTURES and PROPERTIES:
You need to know about functional groups and reactions, and how to apply concepts related to structure such as stereochemistry, acidity/basicity and reactivity etc.
Qu11:
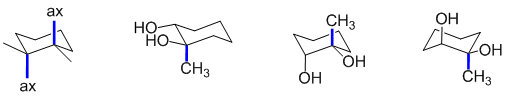
The axial positions are those that point away from the average plane of the ring. In the structures in the question, this means up or down. The structures with axial methyl groups are shown with the axial methyl groups indicated.
Qu12:
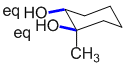
In order for groups to be trans it means that they need to be attached to opposite faces of the ring. Gauche requires them to be at 60 degrees, so we need to look for the diequatorial -OH groups.
Qu13:
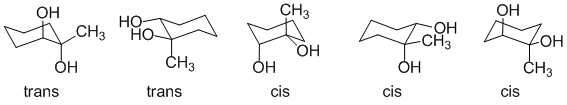
1-methylcyclohexene reacts with cold alkaline potassium permanganate to give a 1,2-diol with syn addition (i.e. cis 1,2-diol):
Qu14:
1-methylcyclohexene reacts with peracid then H3O+ to give a 1,2-diol with anti addition (i.e. trans 1,2-diol):
Qu15:
Best approach is to assign the R/S configurations based on the Cahn-Ingold-Prelog rules. You need to find 2 structures with the same configurations but different conformations.
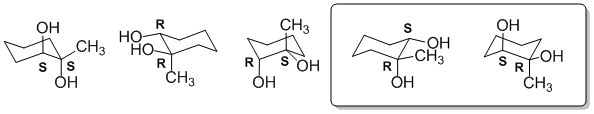
Qu16:
Lowest pKa will be the most acidic.... terminal alkynes are about 25, alkenes about 45 but 1,3-cyclopentadiene is about 16 (due to the aromatic stabilisation of the conjugate base)
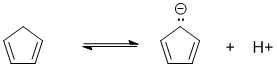
Qu17:
Resonance delocalisation stabilises carbocations. More resonance, more delocalisation implies greater stability:
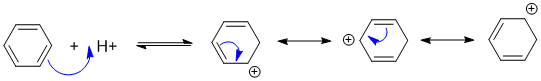
Qu18:
Remember that H on the same C don't have to be equivalent, for example, those on alkenes can be cis or trans to a group....
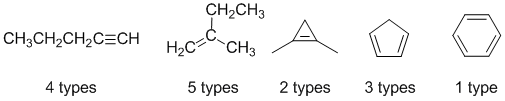
Qu19:
Cyclopentane is C5H10 with an IHD = 1. The only structure of the five with 5 C and an IHD of 1 is the simple alkene.
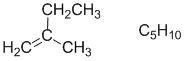
AROMATICITY and RESONANCE:
Best method is to work through the molecules and decide on the aromaticity based on the four criteria for the pi system (cyclic, planar, conjugated pi system with 4n+2 pi electrons).
Qu20:
Conjugation would require a minimum of 2 C=C connected in series.
Qu21:
To be non-aromatic as drawn, we need to find a system that fails on one of the first three criteria (i.e. it lacks a cyclic conjugated pi system) but has a resonance contributor that satisfies all four of the criteria for aromaticity (i.e. it has a pi system that is cyclic, planar, fully conjugated and contains 4n+2 pi electrons). The most likely scenario will involve an exocyclic alkene unit:

Qu22:
To be non-aromatic as drawn, we need to find a system that fails on one of the first three criteria (i.e. it lacks a cyclic conjugated pi system) but has a conjugate base that satisfies all four of the criteria for aromaticity (i.e. it has a pi system that is cyclic, planar, fully conjugated and contains 4n+2 pi electrons). The most likely scenario will involve loss of H+ from a sp3 center to create a conjugated lone pair (so adding 2 electrons to the pi system):
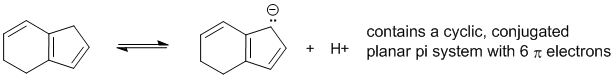
Qu23:
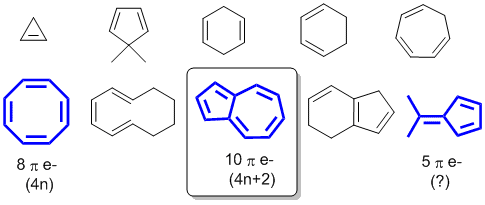
To be aromatic as drawn, we need to find a system that satisfies all four of the criteria for aromaticity (i.e. it has a pi system that is cyclic, planar, fully conjugated and contains 4n+2 pi electrons). Most of the systems don't have cyclic conjugated pi systems, only the 3 shown in blue do. Then the pi electron count is used to establish the aromaticity....
Qu24:
Resonance energy is the stabilisation that results from the interaction of pi systems that are conjugated. The compounds with the no resonance energy would need to be non-conjugated.
Qu25:
In order for the N to be sp2 hybridised it either needs to be part of a double bond or have a lone pair and be next to a pi system.
The non-aromatic systems don't have cyclic conjugated pi systems:
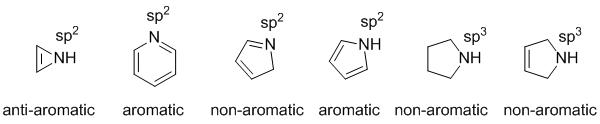
Qu26:
To be aromatic as drawn but with a non-aromatic conjugate acid, we need to find a system that is aromatic as drawn yet when it reacts with H+ is no longer aromatic. A typical example would be where a lone pair of a heteratom is initially part of the aromatic pi system but those electrons are used to make the new bond to the H+.
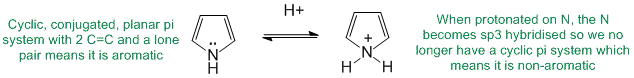
Qu27:
To be anti-aromatic as drawn, we need to find a system that satisfies the first three of the criteria for aromaticity (i.e. it has a pi system that is cyclic, planar, fully conjugated) but contains 4n pi electrons(even number of pi electron pairs)).
Qu28:
To be aromatic as drawn and with a non-aromatic conjugate acid, we need to find a system that has electrons available to interact with the H+ that do not impact the aromaticity.
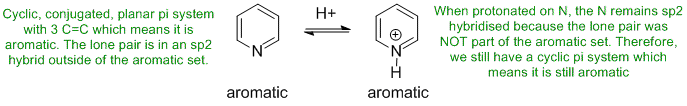
Qu29:
The most common form of tautomerism switches H and double bond positions...
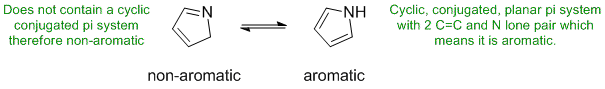
STARTING MATERIALS AND PRODUCTS:
If you are trying to find the product, then you should probably just work forwards through the sequence of reactions.
If you are looking for the starting material, then working backwards is probably the best way to go....
Basically depends on the need to know and identify the reactions, this is often triggered by looking at the functional groups in the molecules.
Qu30:
Working backwards,
the chromium salt looks to have created the ketone from a secondary alcohol. The excess Grignard could have added up to 3 methyl groups based on 2 carbonyl groups (one aldehyde, one ester). The peracid would have made the ester from the ketone (Baeyer-Villager reaction) which was created from the oxidation of the secondary alcohol formed by the anti-Markovnikov hydration of an alkene.

Qu31:
Working forwards, the alkene undergoes syn-dihydroxylation to give a 1,2-diol. The diol is then reacted with a ketone to form a cyclic acetal then the primary alcohol is oxidised with PCC to give the aldehyde. Make sure to pay attention to the stereochemistry of the alkene and hence the diol all the way through...
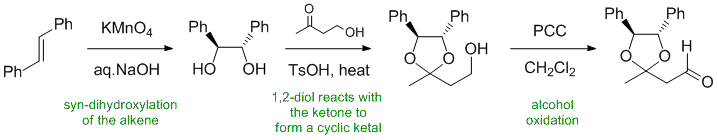
Qu32:
Working forwards, addition of HBr across the alkene in a Markovnikov fashion followed by E2 elimination to give the Zaitsev alkene (more highly substituted). Ozonolysis with an oxidative work up gives a ketone and a carboxylic acid. The ketone is then reduced with the sodium borohydride (but it does not reduce the carboxylic acid).
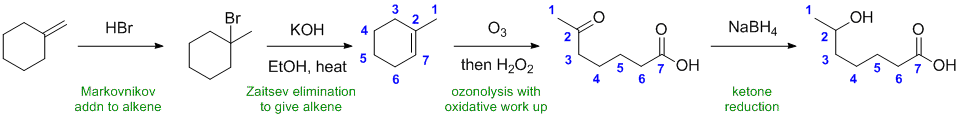
Qu33:
Working backwards... the product is a cyclic ester likely formed via an intramolecular trans-esterification. The crux is managing the stereochemistry based on a wedge-hash product and starting materials that are Fischer projections. However, it's not quite as complex as it might appear because the Fischer projection lends itself to forming rings (groups at are on opposite faces of the ring will be on opposite sides of the Fischer projection) although we have to be careful with the terminal -OH group. So the best way to tackle this is to start with the product and reverse the esterification (red line) to reveal the carboxylic acid and the alcohol group. Then convert this to a Fischer type projection (note that you can get the Fischer projection by realising that groups that are down (hash) in the way the ring was drawn will be on the left of the Fischer projection) and then deal with the terminal -OH group.
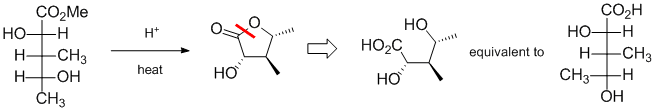
Qu34:
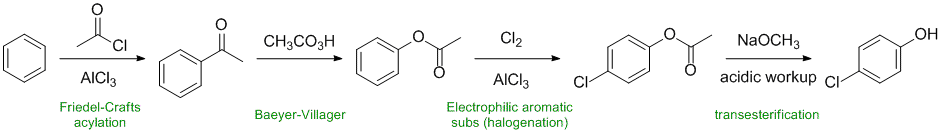
Working forwards....Friedel-Crafts
acylation gives the methyl ketone. The Baeyer-Villager reaction converts the ketone to an ester on the aromatic side. Electrophilic aromatic substitution adds the -Cl being directed o,p by the electron donating group (sterics favour p). The ester is cleaved to leave the phenol.
Qu35:
Working forwards.....Friedel-Crafts
acylation gives the cyclic ketone which is then reduced to the secondary alcohol by the hydride reagent. Dehydration readily yields the conjugated alkene. Radical halogenation is followed by E2 elimination (strong base and heat) to give the conjugated, fully aromatic system.
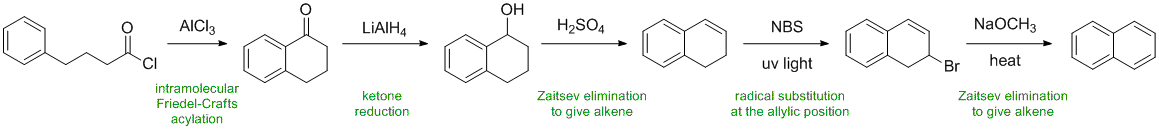
Qu36:
Working forwards....the conjugated diene and the substituted alkyne undergo a Diels-Alder reaction when heated to give the substituted cyclohexadiene. Catalytic hydrogenation reduces both alkenes and sets up the cis stereochemistry of the ester groups. Hydride reduction of the esters gives the primary alcohols with the same stereochemistry.
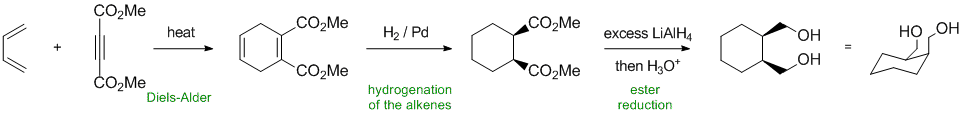
Qu37:
Working backwards....The product ester has been formed from the acyl chloride and an alcohol. The alcohol has been created by the reaction of a Grignard reagent with an epoxide (reacts at the least hindered end) which in turn had been synthesised from the alkene via a 1,2-halohydrin with a base to form an epoxide via an SN2 reaction.
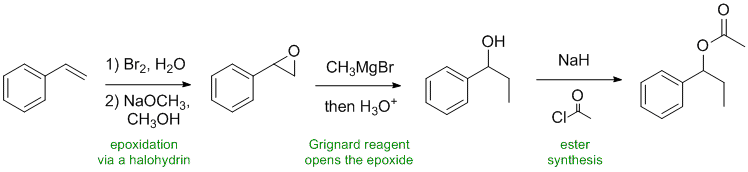
Qu38:
Working backwards.... The most obvious feature is the Wittig reaction to form the alkene by the reaction of the ylide with a ketone. The -OH would have been a problem so there are protection / deprotection steps:
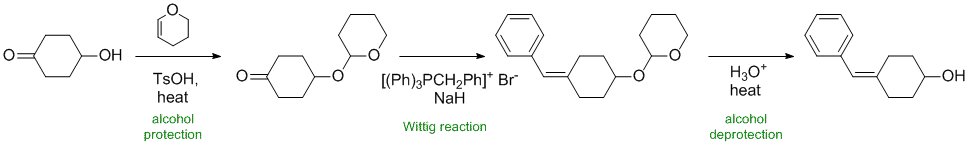
REAGENTS FOR SYNTHESIS:
Need to be able to look at reactions, looking at the functional groups in the starting materials and products of each step to think about
how you have got there.
How well do you know your reagents
? Look at what has actually happened in terms of the reaction functional
group transformation and then first look for any regiochemical issues
then finally the stereochemistry last (it's the hardest to sort out).
Qu39:
To prepare aniline from benzene you need to nitrate then reduce the -NO2 so : i. HNO3 / H2SO4 ii. Sn / HCl iii. NaOH
Qu40:
First you need to analyse the stereochemistry of the Fischer projection and use that to work out if you need to do a syn or anti addition to a cis or trans C=C. That analysis shows that we need to do either a syn addition to a cis-alkene or a anti addition to a trans-alkene. But addition of Cl2 is an anti addition so we must have the trans-alkene and therefore need to use i. Na /NH3 then ii. Cl2.
Qu41:
Need to alkylate the terminal alkyne adding two carbon atoms and then reduce to the cis alkene so use i NaNH2, ii. CH3CH2Br, iii. H2 / Lindlar's catalyst
Qu42:
To rearrange the alkene to a less stable position (less highly substituted and exocyclic) will require adding a leaving group and then forcing an anti-Zaitsev elimination. So add HBr then follow this with an E2 elimination.
Qu43:
Looking at product we need to get the Br at the less highly substituted position (anti-Markovnikov) and syn to the methyl group. HBr / dark gives the wrong regiochemistry and HBr (radical) does not control the stereochemistry. So, add -OH using hydroboration (anti-Markovnikov) and then SN2 to convert the -OH to the -Br and the required stereochemistry.
Qu44:
First you need to analyse the stereochemistry of the Newman projection and use that to work out if you need to do a syn or anti addition to a cis or trans C=C. That analysis shows that we need to do either an anti addition to a trans-alkene or a syn addition to a cis-alkene. Na/NH3 gives trans-alkenes, H2 / Pd will reduce to an alkane and H2 / Lindlar's catalyst gives cis-alkenes. KMnO4 / aq. NaOH results in a 1,2-diol via a syn addition and peracid / H3O+ a 1,2-diol via overall anti addition. Therefore, use i. H2 / Lindlar's catalyst ii. KMnO4 / aq. NaOH / 0 oC.
EXPLANATION OF PHENOMENA
Qu45:
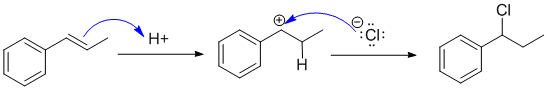
The reaction is the addition of HCl to an alkene which is controlled by the stability of the carbocation intermediate produced when H+ adds to the C=C. In this case favouring a resonance stabilised benzylic carbocation. This means the product is the benzylic chloride. Since there are the same number of groups attached to each end of the alkene, Markovnikov's empirical rule doesn't help us here.
Qu46:
It's the group on the ring that directs the substitution, deactivators direct meta. The methoxy group, -OCH3, is strongly activating and a o,p-director. In most cases, steric effects (size) cause p yields to be higher than o.
Qu47:
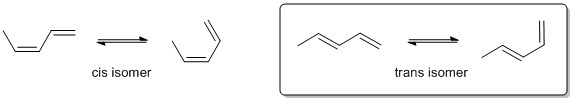
The Diels-Alder reaction requires that the diene adopt the s-cis conformation which means
we need to redraw the systems in that conformation and then assess their stablilities. Steric effects destabilise the s-cis isomer in the s-cis conformation because the methyl group at C1 and the cis H at C4 are close to each other.
Qu48:
Hydroboration / oxidation of terminal alkynes favours the anti-Markovnikov orientation which puts the electrophilic and larger (than H) B atom at the terminal C.
Qu49:
The overview of the reactions is shown below. The analogous product of the alkyne reaction would be the structure shown which would be an anti-aromatic 4 pi electron system.
