Here is an post-mortem analysis / "how to" for the MT. The questions are split by the sections. At the start of each section are a few suggestions of what to look for or how to tackle the question type.
RELATIVE PROPERTIES:
Identify the controlling feature, which is not
always as obvious as it may appear. Look for two pairs of similar systems to compare
that have minimal differences in structure. If a compound is named, draw it out.
If a reaction is involved, identify the type of reaction and then what the controlling
factors are.
Qu1: E
Carbocation
stability.... due to (a) alkyl groups, which are weak electrons donors, (b) resonance with pi bonds and (c) the effect of an oxygen. These effects add stability due to charge delocalisation. i is secondary and doubly allylic, ii is primary and iii like i except it can be further stablised by the lone pairs on the oxygen. This means
that the simple primary cation in i is the least stable of the three.
The oxygen lone pair donation in iii makes it more stable than i. Hence iii > i > ii.
Qu2: C
Configurational isomers are stereoisomers : so look for stereocenters such as chirality centers (e.g. sp3 C with 4 different groups attached). The maximum number of configurational isomers is given by 2n where n is the number of stereocenters. i has two chirality centers (the C with the OH groups) but they are the same sets of four, so we get 3 configurational isomers, (R,R), (S,S) and (R,S). ii has two chirality centers (the C with the OH group and Br groups) so we get 4 configurational isomers, (R,R), (S,S), (S,R) and (R,S). i has two chirality centers (the C with the OH groups) but they are the same sets of four, so we get 3 configurational isomers, (R,R), (S,S) and (R,S). iii has one chirality center (the C with the OH group) so we get 2 configurational isomers, (R) and (S). Note that you can not get cis and trans C=C in a 6 membered ring.
Qu3: E
The reaction is the epoxidation of alkenes. The reactivity towards reduction is determined by the nucleophilicity
of the pi bonds and this is controlled by the number of alkyl groups attached to the C=C unit. i is trisubstituted, ii is disubstituted and iii is tetrasubstituted. So in reactivity, iii > i > ii.
Qu4: C
The reaction is the hydroboration / oxidation of alkenes. First draw out the starting material. i and ii have correctly added the HO and H across the C=C therefore iii has the lowest yield. i is the Markovnikov product, while ii is the anti-Markovnikov product. Since hydroboration / oxidation gives the anti-Markovnikov product as the major product then we get ii > i > iii.
Qu5: E
All about acidity of different types of C-H bonds. i is a vinyl C-H (sp2C-H) and ii is an alkyl C-H (sp3C-H) and iii is an allylic C-H. Acidity is best handled by looking at the reaction HA <=> H+ and A- and look at the stability of A-. The allylic system will be resonance stablised (an allylic anion) and therefore iii is the most acidic here. The greater s character of the sp2 system over the sp3 system makes the vinyl C-H i more stable than the alkyl C-H ii. So in terms of acidity, iii > i > ii.
Qu6: C
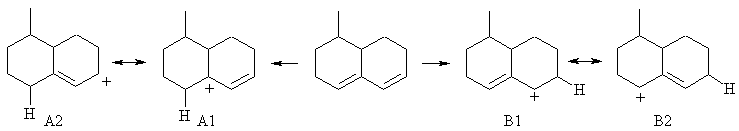
The reaction is the addition of hydrogen bromide to a conjugated diene and here we are under thermodynamic control. Consider the protonations that gives the allylic carbocations A1 abd B1 and then the direct and conjugate addition modes via the alternate resonance forms of those carbocations A2 and B2, then consider the stabilities of those products. i results from direct attack on carbocation A1, ii from conjugate addition via carbocation A2 and iii from the direct addition on carbocation B1. Carbocation A1 is favoured as it is tertiary and allylic, thus the thermodynamic product ii (the trisubstituted alkene) from the A cations is the major product followed by i since it is also formed from the A cations.
Qu7: D
The reaction is the catalytic
hydrogenation reduction of alkenes. The solve the question look at the possible locations where the C=C could have been and pay attention to the symmetry.
Qu8: C
Reaction
is about additions of HX to an alkene. This gives the Markovnikov product
via the most stable carbocation intermediate. The rate of the reaction
is controlled by the formation of the carbocation and the variable here is the
acid. The stronger the acid the faster the rate of carbocation formation. In
terms of acidity HI > HCl > HF (acidity increases down a group in
the periodic table and comparing HX acids to a simple carboxylic acids (pKas
are -9, -7 and 3 respectively). So ii
> i > iii
Qu9: C
Reaction is the Diels-Alder
reaction and we are looking at the dienes reacting with methyl acrylate (the dienophile)....
The reactivity increases in the normal Diels-Alder reaction with electron donating
groups on the diene. The variable here is the groups on the thiophene system. i is unsubstituted, ii has
two alkyl group (a weak electron donor), and iii has electron withdrawing carbonyls in
the form of a ketone. Therefore the reactivity is ii > i > iii.
Qu10: A
Resonance energies measure stability. Here we are looking at different types of polyenes. The longer the conjugated system, the greater the resonance energy. i is has a conjugated triene, ii has a conjugated diene and iii has four isolated C=C units. Therefore we get i > ii > iii.
STARTING MATERIALS AND PRODUCTS:
If you are trying to find the product, then
you should probably just work forwards through the sequence of reactions.
If you are looking for the starting material,
then working backwards is probably the best way to go....
Basically depends on the need to know and identify
the reactions, this is often triggered by looking at the functional groups in
the molecules.
Qu11: D
Working forwards...Alkenes react with acids to give carbocations. If there isn't a very good nucleophile present, then the carbocation can deprotonate to recreate the C=C. When possible, this C=C will tend to be more stable C=C (more substituted C=C) so that the C=C could have rearranged (this situation relates to the Zaitsev rule in the acid catalysed dehydration of alcohols). In this question, the original alkene can protonate to give a secondary carbocation that then undergoes a 1,2-methyl shift to give the tertiary carbocation which then deprotonates to give the tetrasubstituted alkene D. B and E can be rejected because they have pentavalent C atoms.
Qu12: D
Working forwards...E2 elimination of the alkyl halide generates an alkene followed by ozonolysis and a reductive work-up.... should lead to a methyl ketone and aldehyde system, D.
Qu13: AB
Working forwards...bromination of the alkene gives the 1,2-dibromide followed by double elimination (- 2 HBr) with the base will give the conjugated diene. The original location of the C=C dictates the structure. Both A and B will be formed.
Qu14: A
Working forwards...Reaction of an alkene
with a hypohalous gives a 1,2-halohydrin via a cyclic halonium ion. Subsequent reaction with the base will form the epoxide A via an intramolecular Williamson ether synthesis type of reaction.
Qu15: D
Working forwards... The benzyl bromide will undergo SN2 with the nucleophilic terminal acetylide. This is repeated by using the NaNH2 then isopropyl iodide to make an unsymmetrical alkyne which then under goes partial catalytic hydrogenation to the cis-alkene, D.
Qu16: A
Working forwards... The reaction is the hydroboration-oxidation
of an alkene. This syn addition is followed by converting the anti-Markovnikov alcohol into the tosylate and then an E2 elimination. The E2 is forced to be anti-Zaitsev (A) rather than Zaitsev (C) by the trans nature of the TsO leaving group and the adjacent methyl group.
Qu17: B
Working backwards.... look at the product (note it's a substituted cyclic structure
with quite specific stereochemistry... may be a Diels-Alder
reaction? . The second set of reagents indicate ozonolysis with an oxidative work-up, so that explains where the
C=C and the cyclohexene has gone. Once you have the cyclohexene, push the curly
arrows backwards to reveal the diene. Only dienes A and B make
sense. C is not a diene, D has too many C atoms and is not a conjugated diene and E is confused the diene and dienophile components. To decide on A or B then look at the regiochemistry, since the two methyl groups need to lead to the methyl groups of the ketone i.e.B they are beta not alpha to the S.
Qu18: B
See above in Qu 17..... The dienophile is an ethyl ester with a trans methyl group....i.e.B
REGIO- and STEREOCHEMISTRY:
How well do you know your reagents ? Look
at what has actually happened in terms of the reaction functional group transformation
and then first look for any regiochemical issues then finally the stereochemistry
last (it's the hardest to sort out). In cases where more than one product
is formed in equal amounts (e.g. the enantiomers), then both must be selected
for full marks, part marks are given when only one of the pair is selected.
Advice : in each case draw the starting material in the
conformation in which is reacts or the product in the conformation in which
it is initially formed using wedge-hash diagrams. It is a good idea to draw
the materials in such a way that the new bonds are in the plane of the page.
Once you have drawn the materials It may also be good for you to use model kits
for these questions too. Once you have drawn the materials in this way,
you may need to consider rotations around sigma bonds to make your answer match
the options. An alternative approach could be to assign configurations
to your drawn answer to compare them with the options - this can be slow and
prone to error.
Qu19: D
Reaction of an alkene
with a halogen in the presence of water gives a 1,2-halohydrin via a cyclic
bromonium ion, the regiochemistry will put the -OH at the more substituted
position with anti stereochemistry to the Br atom and the -OH group. Treatment with sodium carbonate as a base forms the epoxide with the same stereochemistry as the original alkene. Ring opening of the epoxide under acidic conditions makes the nucleophile MeOH attack the tertiary end of the epoxide with the MeO and OH being anti with respect to the original C=C. B has the wrong regiochemistry.A, C and E have the wrong stereochemistry. Darn those Fischer projections!
Qu20: A
The reagents indicate the catalytic
hydrogenation reduction of the alkenes which is a syn addition. The easiest approach is to imagine twisting the front C in the Newman projections to align the added H atoms in that syn conformation then check the alkyl groups positions to ensure that they are cis as required by the starting material. B, C and E have the wrong stereochemistry. D has the wrong regiochemistry
Qu21: D
The first step is osmium
tetroxide reacting with the alkene to give a 1,2-diols via a syn addition. This is followed by the double dehydration
of an alcohol to give the conjugated diene, followed by a Diels-Alder
reaction. A and B have the wrong functional groups. E is missing a C=C since the dienophile in the Diels-Alder here is an alkyne system. C has the ethyl group in a location that requires a rearrangement of the diene system.
Qu22: C
Looking at the products and the reagents it looks like using a peracid to epoxidise an alkene followed by acidic ring opening using methanol (note the -OH and -OMe groups in the product). Since the opening of the epoxide occurs in an anti type fashion, you need to rotate the Newman projection to put the -OH and -OMe at 180 degrees. With this done, the position of the alkyl groups (t-butyl and phenyl) on the alkene becomes apparent - they are trans. A has the wrong stereochemistry, B and D have the wrong substituents and E has the wrong functional group.
Qu23: AE
Looking at the products and the reagents it looks like the dehydration
of an alcohol followed by anti-Markovnikov addition of hydrogen
bromide to the alkene of 1-ethylcyclohexene - there are several ways to have got to this....A and E can undergo carbocation rearrangements to give the required alkene. We can rule out B and C. We can also rule out D because it only has 7 C atoms.
Qu24: D
Looking at the products and the reagents it looks like the cyclopropanation via a Simmonds-Smith reaction on a trans alkene from a indicate
a dissolving
metal reduction of an alkyne prepared by the alkylation of a terminal acetylide. This means for the a terminal alkyne (A or D) with an isopropyl group.
Qu25: BD
Looking at the products and the reagents it looks like the hydroboration-oxidation
of an alkene where the alkene is cis because of the catalytic
hydrogenation reduction of alkyne to an alkene. The alcohol from the hydroboration is the anti-Markovnikov product so the -OH will be at the least hindered (-methyl) end of the alkene (B or D). Both B and D could have been formed since we can't tell which H in the CH2 adjacent to the cyclohexyl group came from the hydroboration.
PI SYSTEMS:
All about knowing properties of pi systems such as resonance, structure, bonding, shapes, and terminologies etc.
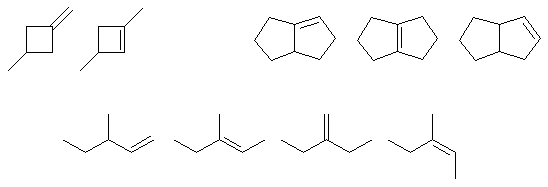
Qu26: ABCD
Conjugation requires an extended pi system (at least 3 atoms) where the pi systems can interact
(parallel, not perpendicular). This is not restricted to all carbon systems.
Make sure you draw out all the bonds to be sure. A has 2 conjugated C=C. B has a empty p associated with the C+ conjugated with the C=C. C has an conjugated alkene and alkyne. D has a C=O in conjugation with a C=C. E is a cumulated diene (pi systems at 90 degrees).
Qu27: AC
Resonance contributors are due to the delocalisation of the pi electrons with the pi system. Here we have a conjugated enone. B and E can be ruled out because they have a different overall charge compared to the original neutral structure. Remember that sp3 centers can't be part of the resonance system.

Qu28: BC
Tautomers (remember alkyne hydration ?) of ketones are enols. A is a resonance contributor. D is a constitutional isomer. E is a conjugate base of the enol of 2-pentanone.
Qu29: BCE
This question brings some laboratory aspects in with lectures, and some
nomenclature. Do you know the structures you worked with?
Sucrose is a carbohydrate, no C=C there. Biodiesel was made from vegetable oil which means unsaturated fatty acids are present and hence C=C. Terephthalic acid and benzoic acid both have benzene rings and hence C=C. Nylon, a polyamide, contain C=O not C=C.
Qu30: D
Allylic hydrogens are those on the carbon atoms adjacent to alkene C=C bonds, i.e. H-C-C=C. A has 4, B none, C none (but it has 12 benzylic), D has 9 and E has 2.
Qu31: C
Polyene stability.... all dienes. Stabilising factors are conjugation, degree of C=C substitution, trans / cis and s-trans / s-cis geometries. C is the most stable because it's a conjugated diene, the C=C are both trans-disubstituted and the C-C connecting the C=C units is s-trans.
Qu32: B
Polyene stability.... Here we are comparing different diene types and we have small ring cycloalkene. Conjugated dienes (A and B) are more stable than cumulated dienes (D). Alkynes (C) are similar in stability to cumulated systems and so an alkyne is less stable than isolated or conjugated dienes. A small ring cycloalkene like E is destablised due to high ring strain. Comparing A and B, the s-trans is more stable than s-cis due to steric effects so B is more stable than A.
Qu33: C
All the bonds are CC bonds, but the hybridisation of the two C involved is changing.
In A, B and E we have sp3 : sp2 C. In D we have a standard sp3 : sp3 CC single bond (154 pm). In C both C are sp2, but we are looking at a C-C
between two C=C in a conjugated diene (about 146 pm).
Qu34: B
The reaction is the hydration
of an alkene to give the Markovnikov alcohol. This reaction is controlled by the formation of the carbocation so the system that gives the most stable carbocation will react the fastest. That will be the tertiary carbocation from B.
![[Chem 353 Home]](../mol.gif)