353 Winter 2012 FINAL
Here is an post-mortem analysis / "how to" for this exam. The questions are split by the sections. At the start of each section are a few suggestions of what to look for or how to tackle the question type.
RELATIVE
PROPERTIES:
Identify the controlling feature, which is not always as obvious as it may
appear. Look for two pairs of similar systems to compare that have minimal
differences in structure. If a compound is named, draw it out. If a reaction
is involved, identify the type of reaction and then what the controlling factors
are.
Qu1: C
Acidity...
if you know your pKa's then this is easy : aldehyde = 17, amine = 35 and ketoester =11. Remember the lower the pKa the stronger
the acid, so ii > i > iii.
What if you don't remember your pKas ? (why not ?) Then you'll
need to deduce it. Think
of the simple acid equation HA <=> H+ A-
then look for factors that stabilise A-.....Note that two of the systems have
the acidic H that are alpha to a carbonyl group. In the aldehyde i, and the ketone and the ester iii, the conjugate bases (they are enolates) can be resonance stablised by the C=O group. The difference between each carbonyl system is the number of carbonyl groups stabilising the enolate. Two carbonyls are better than one. Carbonyl systems are most acidic than amines because of the resoance delocalisation from C to O, and O is more electronegative than either C or N. So ii > iii >i.
Qu2: B
The reaction is electrophilic aromatic substitution, a bromination, and we need to look at the substituent
effects on the aromatic ring. The methoxy group in i is a strong electron donating group via resonance donation of the O lone pair - it's an activating
group. The ammonium group in ii is strongly deactivating due to inductive effects. The methyl group in iii is a slightly activating group due to hyperconjugation. So the reactivity is i > iii > ii.
Qu3: B
The reactivity of carboxylic acid derivative carbonyl groups towards hydrolysis looking at the relative reactivity of i acid anhydride, ii amide,
and iii ester.
The electronic factors of each of the substituents / leaving group on each of the carbonyl groups need to be considered. In i the carboxyl group is a moderate electron donor and a reasonable leaving group. In ii the NH2 is a strong electron donating group and a very poor leaving group. In iii the methoxy group is a strong electron donor and a poor leaving group. Electron donors on the carbonyl make the carbon less electrophilic and less reactive, a better leaving group makes the acid derivative more reactive. Hence
in terms of reactivity i > iii > ii.
Qu4: C
This question is about the E2 reactions of alkyl halides. The key issues are a. the stereochemical prefences for the LG (Br) and the H to be at 180 degrees (i.e. anti) and b. the preference of alkyl groups to be equatorial on cyclohexanes. In i the ring flip to put the Br in the required axial position is feasible and the elimination can occur. This means that in ii when the t-Bu is equatorial, the Br is already axial and readily eliminates, no ring flip required. In iii when the t-Bu is equatorial, so is the Br and because the ring flip is difficult, the elimination is very slow. So, in terms of rate of reaction ii > i > iii.
Qu5: B
First identify the alpha
positions. i is CH3C(=O)CH2R so it has a total of 5H in the alpha postions. ii is an aromatic aldehyde, ArCHO, meaning there are no total enolisable H. iii is an ester with a CH3 group attached to the C=O and
hence 3 enolisable H,
so i > iii > ii.
Qu6: A
The systems are substituted alkenes so we are looking at electrophilic
addition to an alkene . A closer look at the structures shows that the three systems have similar base structures (i.e. alkenes) each with a different substituent: i has an alkoxy group, ii has an alkyl group and iii a trifluoromethyl group. Each alkene will react with the acid to give a carbocation and the substituent will affect the availability of the pi electrons: electron donating substituents will make them more available and electron withdrawing will make them less available. An alkoxy group,
RO-, in i is a strong electron donor, an alkyl group, -R, in ii is a weak electron donor and a CF3 group is a strong electron withdrawing. So i will be
more reactive than ii (think of the electron donating alkoxy group making
the C=C more electron rich) and iii the least reactive... so i > ii >iii.
Qu7: D
Acidity...
if you know your pKa's then this is easy (after you locate the most acidic H in each): ketone = 20, carboxylic acid = 5 and ketoester =11. Remember the lower the pKa the stronger
the acid, so in terms of acidity ii > iii > i.
What if you don't remember your pKas ? (why not ?) Then you'll
need to deduce it. Think
of the simple acid equation HA <=> H+ A-
then look for factors that stabilise A-.....Note that two of the systems have
the acidic H that are on C alpha to a carbonyl group. i and iii. In i, note that it's the methl groups that are relevant and the H there are only next to one C=O. In the ketone and the ester system iii, the conjugate bases can be resonance stablised by both of the C=O groups. The difference between each carbonyl system is the number of carbonyl groups stabilising the enolate. Two carbonyls are better than one. What about ii ? It's a carboxylic acid, so the most acidic H is attached to O and the charge can be delocalised to a second electronegative O atom. ii > iii > i.
Qu8: E
The reaction is electrophilic aromatic substitution, a nitration and we need to look at the substituent
effects on the aromatic ring. The alkyl groups in i and iii are slightly electron donating groups via hyperconjugation - it's are activating
group and ortho/para directing. The tert-butyl group in iii is large - it's steric size will favour para since the ortho groups will be partially blocked. The trichloromethyl group in ii is strongly deactivating due to inductive effects, and is meta directing group. So the para yield will be iii > i > ii.
Qu9: D
Acidity...
Think
of the simple acid equation HA <=> H+ A-
then look for factors that stabilise A-.....All are phenols, all that is changing is the substituent on the benzene ring. Electron donating substitutents on the ring will destablise the phenolate (the conjugate base A-). The alkoxy group, RO-, in i is a strong electron donating group via resonance donation of the O lone pair. ii has an nitro group, -NO2, which is a strong electron withdrawing group and iii is just H, our reference group (i.e. no electronic effect). So the acidity is ii > iii > i.
Qu10: C
The reactivity of carbonyl groups towards hydride from NaBH4, depends on the electrophilicity of the carbonyl carbon. Looking at the carbonyls, we have three aldehydes, the difference is the substituent on the aromatic ring. An electron withdrawing group will make the C in the C=O more electrophilic and therefore more reactive. In i our substituent is just H, our reference group (i.e. no electronic effect). ii has an nitro group, -NO2, which is a strong electron withdrawing group. iii has an allkoxy group, RO-, a strong electron donating group via resonance donation of the O lone pair. So overall, ii > i > iii.
STRUCTURES and PROPERTIES:
You need to know about functional groups and reactions, and how to apply concepts
related to structure such as stereochemistry, acidity/basicity and reactivity etc.
Qu11: any 2 of BCE
The axial positions are those perpendicular to the plane of the ring rather than around the average plane of the ring (equatorial).
Qu12: B
For the -OH groups to be trans, they need to be on opposite faces of the ring (as in A and B). The equatorial positions are those around the plane of the ring.
Qu13: any 2 of CDE
1-methylcyclohexene will react with cold alkaline KMnO4 to give the 1,2-diol via a syn-addition - this means that the two -OH groups are on same face of the ring i.e. cis. The relative positions of the -OH groups are trans in A and B, and cis in C, D and E.
Qu14: A or B
The opposite to qu 13.
N atoms that part of one double bond will usually be sp2 hybridised (B and E). Neutral N atoms where the N lone pair is interacting with an adjacent pi system (i.e. resonance) will be sp2 to allow the lone pair to be in a p orbital (C and D).
Qu16: BDE
Protonation of either B or E will give N+ where the N is also part of a double bond so those N are (still) sp2 hybridised. A and C will protonate to give N with 2 alkyl groups and 2 H atoms attached, and hence 4 groups and sp3 hybridised. Amides (B) protonate on O (draw the resonance contributors to check this out!), and then means the N is still sp2.
Qu17: B
The lowest pKa means the most acidic. Knowing pKas will help, if not knowing the factors that affect acidity is vital. First note that all the systems are C-H bonds, the characteristics of the C are changing. C is a terminal alkyne (pKa = 25), D has vinyl and allyl H (pKa about 50 and 43 respectively) and E is has aromatic H (pKa = 43). The key issue here is that cyclopentadiene (B) is non-aromatic and has an aromatic (and hence much more stable) conjugate base, thus, cyclopentadiene is quite acidic (pKa about 16).
Qu18: E
Consider each of the carbocations produced when each system reacts with H+. E gives a carbocation that is conjugated with a conjugated diene which means the carbocation is the most resonance stabilised (i.e. has the most resonance contributors).
Qu19: D
A has two types of H (sp3 CH2 and sp2 CH), B has three types (sp3 CH2 and 2 x sp2 CH), C has two types of H (sp3 CH3 and sp CH), D has four types (sp3 CH2, sp2 CH and cis and trans H in sp2 CH2), E has one type of H (Ar sp2 CH).
Qu20: A
Best derived by pushing arrows. The key issue is that the +ve charge can only be relocated to an atom that was originally part of the pi system, i.e. a C that was not sp3 hybridised in the origianl structure.
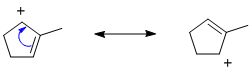
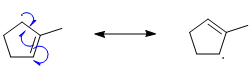
Qu21: A
Best derived by pushing arrows. The key issue is that the single radical electron can only be relocated to an atom that was originally part of the pi system, i.e. a C that was not sp3 hybridised in the origianl structure.
AROMATICITY and RESONANCE:
Best method is to work through the molecules and decide on the aromaticity based on the four criteria (cyclic, planar, conjugated pi system with 4n+2 pi electrons).
Qu22: AD
Only AD has a planar, cyclic and conjugated pi system, and in this case it has 5 pairs of pi electrons (i.e. n = 2 in the Huckel rule).
Qu23: BC
The non-aromatic compounds here are A, B,C, D,E, AB, AC, AE, and BC. Only BC has an aromatic resonance structure (where the exocyclic C=C is broken to make a tertiary carbocation and an anionic cyclopentadiene (which is aromatic where n=1, a 6 pi electron system)).
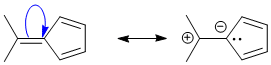
Qu24: AE
The non-aromatic compounds here are A, B,C, D,E, AB, AC, AE, and BC. To form a conjugate base, we remove a proton, H+, to leave an aromatic anion. Only AE has an aromatic conjugate base (where there is an anionic cyclopentadiene (which is aromatic where n=1, a 6 pi electron system)). A and E would form 4n Huckel systems (i.e. even numbers of pairs) implying anti-aroatic type systems.
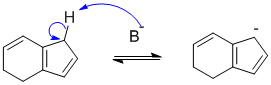
Qu25: C
Polyenes require not or more C=C units (rules out A) and non-conjugated requires that the C=C units be separated by at least one sp3 C. This can only be C.
Qu26: A or C
The systems with the no resonance energy need to be non-conjugated, i.e. requires that the C=C units be separated by at least one sp3 C or that there only be one C=C.
Qu27: AC
The aromatic compounds as drawn here are A, C, E, AB, AC and AD. For the conjugate acid to be non-aromatic it means that adding a proton must create a structure that violates one of the first three aromaticity criteria. The conjugate acids of A, C, E, AB and AD are all still aromatic since there is a lone pair in each that is not part of the pi system.
Qu28: AD
The aromatic compounds as drawn here are A, C, E, AB, AC and AD. The most common form of tautomerism switches H and double bond positions.
![]()
Qu29: AE
The non-aromatic compounds here are D and AE. Remember that atoms can not move in resonance contributors. Here AE has a ylid from where the exocyclic P=C is broken to make a phosphonium ion and an anionic cyclopentadiene (which is aromatic where n=1, a 6 pi electron system)).
Qu30: B
The boron in B has a empty p orbital that creates the cyclic conjugated pi system but since there are no electrons in this orbital, it's a 4 pi system (an even number of pi electron pairs, 4n where n=1).
Qu31: E
The aromatic compounds as drawn here are A, C, E, AB, AC and AD. For n = 2 in the Huckel rule, we require 10 pi electrons. A, C, AB, AC and AD are all 6 pi electron systems.
STARTING MATERIALS AND PRODUCTS:
If you are trying to find the product, then you should probably just work forwards through the sequence of reactions.
If you are looking for the starting material, then working backwards is probably the best way to go....
Basically depends on the need to know and identify the reactions, this is often triggered by looking at the functional groups in the molecules.
Qu32: A
Working forwards, the peracid reacts with the alkene to give an epoxide. The Grignard reagent will then react with the epoxide at the least hindered end to form a tertiary alcohol (A or E). E has the methyl group on the wrong carbon atom.
Qu33: B
Working forwards, hydroboration and oxidation of the alkene gives the anti-Markovnikov primary alcohol. PCC is an oxidising agent giving the aldehyde.
Qu34: A
Working forwards, furan is a diene and maleic anhydride is a dienophile : a Diels-Alder reaction. The anhydride is an acid derivative and will react with EtOH to make an ester. Catalytic hydrogenation will remove the alkene C=C formed in the cyclohexene unit during the Diels-Alder reaction. Since the anhydride is cyclic, it's effectively cis and Diels-Alder reactions tend to be endo selective. B is the exo product, C has come from a trans dienophile. D still has the C=C and E is lacking the O from the furan.
Qu35: B
Working forwards, the first step is an electrophilic
aromatic substitution, specifically Friedel-Crafts
acylation adding an ethyl ketone. The second step is hydride reduction by excess lithium aluminium hydride that reduces the ketone to a secondary alcohol, so B.
Qu36: B
Working forwards, ketones react with peracid to undergo a Baeyer-Villager reaction to give an ester, in this case a cyclic ketone will give a cyclic ester (also known as a lactone). The ester will rect with two equivalents of the Grignard to make a tertiary alcohol from the C=O and a primary alcohol (the ester leaving group). Count the C in the main chain (5).
Qu37:A
Working backwards, in step 2, the 1,2-diol has been formed via basic hydrolysis of the epoxide from step 1, the peracid reacts with the alkene to give an epoxide. The formation of a diol via these reactions is an overall anti addition to the original alkene. Therefore, we need to manipulate the Fischer diagrams into wedge-hash diagrams and then rotate those in to the conformation with the -OH groups anti and hence reveal the alkene (3-methylhexene) and it's stereochemistry. D and E have the wrong alkene structure : no need for the -OH. Bis the wrong alkene and C has the wrong stereochemistry.
Qu38:A
Not an easy one because simple acids and bases are the hardest to work out what they are doing. Anyway, working backwards, step 4 hints at an enolate (not the case) and step 3 the formation of an epoxide from an alkene, step 2 NaBH4 would have reduced the original ketone to a secondary alcohol. Step 1 is the reaction of an enolate of an active methylene compound. Only A makes sense.... the enolate in step 1 will react with the bromide to alkylate the starting material and have the alkene to form the epoxide in step 3, then the base will deprotonate the more acidic alcohol, leading to the formation of an alkoxide that functions as the nucleophile to produce the required product.
Qu39: C
Working backwards, the product is a cyclic ketal formed by the reaction of cyclohexan-1,2-diol with a ketone (propan-2-one). Since we have the ketone in the reagents in step 2 we need to make the 1,2-diol. Looking at the reagents, this has come from the syn-addition to an alkene.
Qu40: E
Working forwards, the first step is the reduction of the diester by the excess hydride to the 1,2-diol. Reaction of the 1,2-diol with the ketone will form a cyclic ketal. The key here is getting the right number of atoms in each ring. A would be formed by reaction with cyclopentanone with 1,3-propanediol. B would be formed by reaction with cyclohexanone with 1,2-ethanediol. C would be formed from an intramolecular system (1,8-dihydroxyheptan-4-one), and D would be formed by reaction with cyclohexanone with 1,3-propanediol.
REAGENTS FOR SYNTHESIS:
Need to be able to look at reactions, looking at the functional groups in the starting materials and products of each step to think about
how you have got there.
How well do you know your reagents
? Look at what has actually happened in terms of the reaction functional
group transformation and then first look for any regiochemical issues
then finally the stereochemistry last (it's the hardest to sort out).
Qu41: BE
Overall we need to achieve the alkylation of an active methylene compound via an enolate. Since there is an ethyl ester, ethoxide is a good choice and we need a C3 chain with a Br AND another leaving group.
Qu42: BC
Counting carbons from the Br suggests we've lost the ester group - that implies ester hydrolysis and decarboxylation best done together by heating the ester with aqueous acid.
Qu43: CE
There has been a reduction of the ketone to a secondary alcohol typically done with hydride reagents.
Qu44: C
Formation of a silyl ether as a protecting group for the alcohol.
Qu45: AD
The bromide has been reacted in an SN2 fashion with a P nucleophile (first step in the Wittig reaction).
Qu46: A
Formation of the ylid from the phosphonium salt requires a strong base such as an alkyl lithium (second step in the Wittig reaction).
Qu47: AB
Reaction of the ylid with the appropriate aldehyde or ketone to make the alkene (third step in the Wittig reaction).
Qu48: DE
Deprotection of the silyl ether group to regenerate the alcohol. Silyl ethers are cleaved using F-.
Qu49: AC
Oxidation of the secondary alcohol to give the ketone.
Qu50: ABC
Conversion of an alkene into a 1,2-diol.
EXPLANATION OF PHENOMENA
Qu51: E
Phenyl ethanoate has an ester group connected through the -O- on the benzene ring i.e. RCO2-and this group is activating and directs ortho/para.
Qu52: D
9-BBN is a sterically hindered hydroboration reagent. It is a combination of electronic and steric effects that cause the electrophilic B atom to attach to the least substituted (least hindered) end of the alkyne.
Qu53: D
The reactivity of carbonyl groups towards nucleophiles depends on the electrophilicity of the carbonyl carbon which is controlled by the electronic effects of the attached groups. In the carbonyl group, the C is electrophilic due to the attached electronegative O atom. If the substituent if electron donating, it will decrease the electrophilicity and therefore the reactivity of the C.
Qu54: B
When temperature affects product distribution is this way, we typically have a case of kinetic and thermodynamic control. At the lower temperature, the reaction is under kinetic control where it is the rate of reaction to form the enolate intermediate that is important and at higher temperature it is the stability of the enolate that is important.
Qu55: E
Basicity can be addressed by looking at the availability of the pair of electrons. More available = more basic. The key here is the aromatic character of imidazole and that one N is part of a double bond and the other N contributes a lone pair to the pi system.
![[Chem 353 Home]](../mol.gif) Return to Homepage
Return to Homepage