353 Winter 2013 FINAL
Here is an post-mortem analysis / "how to" for this exam. The questions are split by the sections. At the start of each section are a few suggestions of what to look for or how to tackle the question type.
RELATIVE
PROPERTIES:
Identify the controlling feature, which is not always as obvious as it may
appear. Look for two pairs of similar systems to compare that have minimal
differences in structure. If a compound is named, draw it out. If a reaction
is involved, identify the type of reaction and then what the controlling factors
are.
Qu1: E
Acidity...Remember the lower the pKa the stronger
the acid, and think
of the simple acid equation HA <=> H+ A-
then look for factors that stabilise A-.....In all three cases we are looking at C-H systems. In i we have a terminal alkyne, so it's an spC-H bond. The carbanion is stabilised because the -ve charge is assoicated with an sp hybrid orbital and is close to the +ve nucleus. In ii the key issue is that deprotonation gives a highly stable aromatic carbanion. In iii the conjugate base is resonance stablised by the C=C group. The aromatic system in ii is the most significant, followed by the sp effect in the terminal alkyne (if you know your pKa's then this is easy : terminal alkyne about 25, cyclopentadiene about 15 and cyclopentene about 45). So iii > i > ii.
Qu2: B
The reaction is electrophilic aromatic substitution, Friedel-Crafts alkylation, and we need to look at the substituent
effects on the aromatic ring. The methoxy group in i is a strong electron donating group via resonance donation of the O lone pair - it's an activating
group. The cyano group in ii is strongly deactivating due to inductive and resonance effects. The halogen group in iii is a slightly deactivating group due to inductive effects (due to electronegativity). So the reactivity is i > iii > ii.
Qu3: AB
The reactivity of carbonyl groups towards hydride reduction (also relates therefore to one of the laboratory experiments) looking at the relative reactivity of i an ester, ii a ketone,
and iii an aldehyde.
The key factor is how each of the substituents on each of the carbonyl groups affects the electrophilicity of the carbonyl carbon. In i the -OR group is a strong electron donor. In ii the -R group is a weak electron donor. In iii the group is an H and so is aour reference point (i.e. electronically neutral). Electron donors on the carbonyl make the carbon less electrophilic and less reactive. Hence
in terms of reactivity iii > ii > i.
Qu4: A
Resonance energy indicates the stability of the conjugated system. Aromatic systems have high stablisation and therefore high resonance energies. i is benzene which has the most resonance energy (36 kcal/mol, our "model" aromatic compound). ii is furan, which is also an aromatic system (16 kcal/mol), but not as aromatic as benzene. iii is cyclopentadiene which is not aromatic, it's just a conjugated diene (4 kcal/mol) . Hence i > ii > iii.
Qu5: B
First identify the alpha positions. i is CH3C(=O)CH2R so it has a total of 5H in the alpha postions. ii is an aldehyde, CH3CHO, meaning there 3H in the alpha postions. iii is a cyclic ketone with a CH2 group attached to either side of the C=O and
hence 4 enolisable H,
so i > iii > ii.
Qu6: A
The systems are substituted alkenes so we are looking at electrophilic
addition to an alkene . A closer look at the structures shows that the three systems have similar base structures (i.e. alkenes) each with a different substituent: i has an alkoxy group, ii has an alkyl group and iii a nitro group. Each alkene will react with the acid to give a carbocation and the substituent will affect the availability of the pi electrons: electron donating substituents will make them more available and electron withdrawing will make them less available. An alkoxy group,
RO-, in i is a strong electron donor, an alkyl group, -R, in ii is a weak electron donor and a NO2 group is a strong electron withdrawing. So i will be
more reactive than ii (think of the electron donating alkoxy group making
the C=C more electron rich) and iii the least reactive... so i > ii >iii.
Qu7: C
Acidity...
if you know your pKa's then this is easy (after you locate the most acidic H in each): ketone = 20, carboxylic acid = 5 and ester =25. Remember the lower the pKa the stronger
the acid, so in terms of acidity ii > i > iii.
What if you don't remember your pKas ? (why not ?) Then you'll
need to deduce it. Think
of the simple acid equation HA <=> H+ A-
then look for factors that stabilise A-.....Note that two of the systems have
the acidic H that are on C alpha to a carbonyl group (i and iii). In the ketone i and the ester iii, the conjugate bases can be resonance stablised by both of the C=O groups. The difference between each are the different groups adjacent to the C=O and how they stablise or destabilise the enolate. In the ketone, the group is and alkyl group which is a weak electron donor while in the ester the group is an alkoxy group which is a strong electron donor. Electron donors destabilise anions (and therefore enolates). What about ii ? It's a carboxylic acid, so the most acidic H is attached to O and the charge can be delocalised to a second electronegative O atom. So ii > i > iii.
Qu8: A
The reaction is electrophilic aromatic substitution, a bromination and we need to look at the substituent
effects on the aromatic ring. The alkyl group in iii is slightly electron donating groups via hyperconjugation - it's an activating
group and ortho/para directing but the tert-butyl group is large - it's steric size will favour para since the ortho groups will be partially blocked. The halogens in i and ii are weakly deactivating due to inductive effects, but are also ortho/para directing groups, but increasing the steric size will increase the para yield as above. Of these groups, t-butyl is the largest and Br > Cl..... so % ortho product will be i > ii > iii.
Qu9: B
The basicity of phenolates is determined by the electron availability of the electrons on the -ve O atom. This is affected as the substituents change. In this question we have 3 different substituents which stablise the -ve O to different extents, the stronger the withdrawing effect the less basic the O atom is (since the electron withdrawing group decreases the availability of the electrons and this makes the base less able to donate those electrons). In i we have a methoxy group which is an electron donating group. ii has a nitro group which is strongly electron withdrawing group via resonance and inductive effects. The bromine group in iii is a weakly deactivating group due to inductive effects (due to electronegativity). So the basicity is i > iii > ii.
Qu10: E
All the bonds are CH bonds. The factors involved are (1) the hybridisation of the C atom and (2) allylic systems. In terms of hybridisation, sp3 C-H bonds are weaker then sp2 C-H due to the increased s character in the sp2 situation creating a shorter and stronger bond. Allylic C-H bonds are weaker due to the proximity of the electron rich pi bond. In i we have a secondary sp3 system. In ii is secondary sp3 system that is allylic and in iii a vinyl C-H i.e. sp2C.. So in terms of C-H bond strengths: iii > i > ii.
STRUCTURES and PROPERTIES:
You need to know about functional groups and reactions, and how to apply concepts
related to structure such as stereochemistry, acidity/basicity and reactivity etc.
Qu11: A
The most acidic will be the -OH systems.... phenols are more acidic than simple alcohols (due to the extra resonance delocalisation of the conjugate base of phenols).
Qu12: E
Aldehydes and ketones react with water to give hydrates.... E is an aldehyde.
Qu13: B
Ketones reduce to give secondary alcohols.
Qu14: E
The shortest CO bond will be the double bond (note that C has resonance and the interaction with make the 2 CO bonds be between double and sinlge in length).
Phenyl ethanoate is an ester and will hydrolyse to give phenol and ethanoic acid.
Qu16: D
A and C each have 2, B and E each have 1. D has 3 because of the resonance interaction of the N in the amide creates restricted rotation about the CN bond and makes it behave like a double bond and hence cis/trans type stereochemistry applies.
Qu17: ABC
LiAlH4 reduces
aldehydes (A), carboxylic acids, esters (C) and acid anhydrides (B) to primary alcohols. Amides (D) are reduced to amines and ketones (E) to secondary alcohols.
Qu18: D
Lowest pKa = strongest acid. The NH group in an amide has a pKa of about 15. Aldehydes (A) pKa = 17, acid anhydrides (B) are not noted for there acidity, esters (C) pKa = 25, and ketones (E) pKa = 20.
Qu19: B
The reactivity of carboxylic acid derivative carbonyl groups towards hydrolysis depends on the electronic factors of each of the substituents / leaving group on each of the carbonyl groups. The weaker the group is as an electron donor the more electrophilic and the more reactive the C=O. The better the leaving group, the more reactive the system.
AROMATICITY and RESONANCE:
Best method is to work through the molecules and decide on the aromaticity based on the four criteria (cyclic, planar, conjugated pi system with 4n+2 pi electrons).
Qu20: AE
Only AE has a planar, cyclic and conjugated pi system, with 3 pairs of pi electrons (i.e. n = 1 in the Huckel rule).
Qu21: A
The non-aromatic compounds here are A, B, C, E, AB, and AC. Only A has an aromatic resonance structure (where the exocyclic C=C is broken to make a tertiary carbocation at the carbon in the ring that is part of the exocyclic alkene which is aromatic where n=1, a 6 pi electron system). Note the cyclics structures that have an sp3 C in the cycle can not become aromatic due to a resonance interaction (this rules out B, C, E, AB, and AC).
Qu22: A
The non-aromatic compounds here are A, B, C, E, AB, and AC. Addition of H+ to the exocyclic alkene in A will make a tertiary carbocation at the carbon in the ring that is part of the exocyclic alkene which is aromatic where n=1, a 6 pi electron system.
Qu23: A
A tetraene has four C=C (A, AC) of which only A is conjugated.
Qu24: D
As drawn, the non-aromatic compounds here are A, B, C, E, AB, and AC. As drawn the aromatic compounds here are AD, AE and BC. D is anti-aromatic as drawn as it has a planar, cyclic and conjugated pi system, with 4 pairs of pi electrons.
Qu25:D
The most acidic of the systems is expected to be cationic (i.e. one of B, D or AC). D will be more acidic than B or AC because the conjugate base of D gains aromatic stability while B and AC are aromatic (D is non-aromatic).
Qu26: AD
The most basic of the systems is expected to be neutral with the most available lone pairs. N is typically more basic than an analogous O (due to electronegativity). N lone pairs involved in resonance are expected to be less available = less basic. Note that AE is quite basic due to the aromatic nature of the product from O protonation.
Qu27: B or C or AC
The aromatic compounds as drawn here are A, B, C, AB, AC, AE and BC. For the conjugate acid to be non-aromatic it means that adding a proton must create a structure that violates one of the first three aromaticity criteria. The conjugate acids of A, AB, AE and BC are all still aromatic since there is a lone pair in each that is not part of the pi system. C is the most obvious answer.
Qu28: D or AD
In order for the heteroatom (i.e. non C or H atom) to be sp3 hybridised, it can't be involved in resonance.
Qu29: E
The most common form of tautomerism switches H and double bond positions and the involvement of carbonyl groups is quite common:
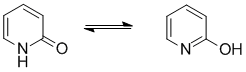
STARTING MATERIALS AND PRODUCTS:
If you are trying to find the product, then you should probably just work forwards through the sequence of reactions.
If you are looking for the starting material, then working backwards is probably the best way to go....
Basically depends on the need to know and identify the reactions, this is often triggered by looking at the functional groups in the molecules.
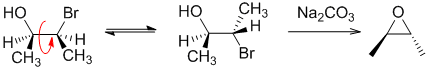
Qu30: E
Working forwards, treatment of a 1,2-halohydrin with sodium carbonate as a base forms the epoxide via an SN2 reaction. This means that we need to set up the starting material in the conformation in which is reacts where the HO and the Br are anti to each other so that the Nu can attack the backside (at 180 degrees to the LG) and cause the normal SN2 inversion. A, B and Call have the wrong functional groups. D has the wrong stereochemistry because you've ignored the SN2 stereochemical requirements.
Qu31: B
Working forwards, the terminal alkyne undergoes hydroboration / oxidation to give the aldehyde. Hydride reduction of an aldehyde gives a primary alcohol.
Qu32: E
Working forwards, a conjugated diene and (di)substituted alkene (the dienophile) .... it's a Diels-Alder reaction. The reagent in step 2 is DIBAL (a selective hydride reducing agent) which will reduce the ester groups to aldehydes (after work up = step 3). Then in step 4, catalytic hydrogenation, reduce the alkenes C=C to an alkane C-C. This is a syn reduction so the product is a cis-1,2-dialdehyde.
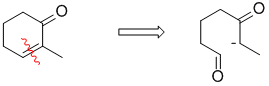
Qu33: C
Working backwards, not an easy one because simple acids and bases are the hardest to work out what they are doing. Anyway, looking at the product (it's a conjugated enone) hints at an intramolecular aldol reaction. The required keto-aldehyde has been obtained via ozonolysis followed by a reductive work up. Looking at the product, and "breaking" the C=C shows that we need an ethyl ketone and an aldehyde which comes from ethylcyclopentene.
Qu34: B
Working forwards, the ketone will undergo a Baeyer-Villager reaction to give the cyclic ester (O inserts on the more substituted side) followed by hydride reduction to give a diol after work up.
Qu35: C
Working backwards, PCC is an oxidising agent giving the ketone from a secondary alcohol formed by the attack of an acetylide on an aldehyde. Hydration of the terminal alkyne gives the other ketone group. The keys to this question are (i) the number of C atoms and (ii) the location of type of alcohol obtained when reacted with an acetylide.
Qu36: D
Working backwards, in step 2, we have formed a Grignard reagent and then reacted it with H+ which overall acheives the conversion of a halogen to a hydrogen. The first step is an electrophilic
aromatic substitution, specifically Friedel-Crafts
alkylation adding an ethyl group. In order for this to be ortho to the Cl, the para position needed to be blocked, here with Br (which is more reactive to Mg than Cl).
Qu37: E
Working backwards, looking at the product, it looks like we have done a Friedel-Crafts
acylation followed by a Wolff-Kischner reduction as a way of adding a linear alkyl group to a benzene. Step 1 is the way to prepare an acyl chloride from a carboxylic acid ready for that Friedel-Crafts acylation.
Qu38: E
Working forwards, the first step is the reaction of a Grignard reagent with a simple aldehyde which after work up gives a secondary alcohol... count carbons.
REAGENTS FOR SYNTHESIS:
Need to be able to look at reactions, looking at the functional groups in the starting materials and products of each step to think about
how you have got there.
How well do you know your reagents
? Look at what has actually happened in terms of the reaction functional
group transformation and then first look for any regiochemical issues
then finally the stereochemistry last (it's the hardest to sort out).
Qu39: CE
Overall we need to achieve the alkylation of an ester via an enolate. Since there is a t-butyl ester, LDA is the best choice.
Qu40: D
Reduce the ester to a primary alcohol which is typically done with a hydride reagent.
Qu41: B
Convert the primary alcohol to a tosylate using tosyl chloride and a base.
Qu42: DE
Reaction of a terminal alkyne with a strong base will give the acetylide ion.
Qu43: ABC
There has been a reduction of the alkyne to a trans-alkene typically done via a dissolving metal reduction.
Qu44: BC
Deprotection of the THP ether to recover the alcohol. Note it's a type of acetal.
Qu45: ABE
Convert an alkene to an expoxide using a peracid.
Qu46: E
Convert the alcohol to an ester by reaction with a carboxylic acid (or more reactive derivative, the anhydride).
Qu47: AD
Convert the alkene to an aldehyde minus 3 C atoms using an ozonolysis with a reductive work up.
Qu48: AC
Oxidise the aldehyde to a carboxylic acid using an aqueous chromium reagent.
Qu49: BE
Convert the carboxlic acid to a methyl ester using a base (here carbonate) to make the nucleophilic carboxylate ion and then SN2 reaction with an alkyl halide.
Qu50: BD
A tough one....need to cleave an ethanoate ester to the alcohol while leaving a methyl ester and an epoxide in tact.... so use a trans-esterification.
EXPLANATION OF PHENOMENA
Qu51: B
Propene reacts with HBr in the presence of peroxides via a radical addition (via the formation of a secondary radical) to give the anti-Markovnikov product, 1-bromopropane.
Qu52: E
The reactivity of carbonyl groups towards nucleophiles depends on the electrophilicity of the carbonyl carbon which is controlled by the electronic or steric effects of the attached groups. The F atoms in hexafluoropropane make the carbonyl more reactive due to the electronegative effects of the F via the sigma bonds (induction).
Qu53: E
The ethoxide ion is a base and reacts with the carboxylic acid to give a carboxylate ion. This is a simple acid / base reaction.
Qu54: D
Electrophilic aromatic substitution is typically controlled by the stability of the intermediate carbocation formed by the attack of the electrophile on the aromatic starting material, e.g. consider the rationale for substituent effects.
Qu55: D
Sodium borohydride will reduce the aldehyde to a primary alcohol which could undergo intramolecular esterification.
![[Chem 353 Home]](../mol.gif) Return to Homepage
Return to Homepage