Here is an post-mortem analysis / "how to" for the Final. The questions are split by the sections. At the start of each section are a few suggestions of what to look for or how to tackle the question type.
RELATIVE PROPERTIES:
Identify the controlling feature, which is not
always as obvious as it may appear. Look for two pairs of similar systems to compare
that have minimal differences in structure. If a compound is named, draw it out.
If a reaction is involved, identify the type of reaction and then what the controlling
factors are.
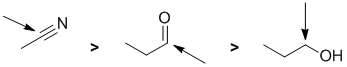
Qu1:
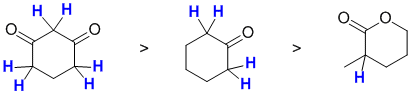
Acidity...Remember the lower the pKa the stronger the acid, and think of the simple acid equation HA <=> H+ A- then look for factors that stabilise A-.....In all three cases we are looking at C-H systems.We have two alkynes, one terminal alkyne and one internal. For the terminal alkyne, the C-H bond is an sp C-H bond. The carbanion is stabilised because the -ve charge is associated with an sp hybrid orbital and is close to the +ve nucleus. In the internal alkyne, deprotonation of an sp3 C-H gives a resonance stabilised carbanion. For cyclopentadiene, the conjugate base is aromatic and the resonance stabilisation by the aromatic system in is the most significant, followed by the sp effect in the terminal alkyne (if you know your pKa's then this is easy : terminal alkyne about 25, internal alkyne (45) and cyclopentadiene about 16). So for strongest acid to weakest acid:
![]()
Qu2:
The reaction is electrophilic aromatic substitution, nitration, and we need to look at the substituent effects on the aromatic ring. The substituents are a bromine, a ketone and an ester. Note that the ester group is attached through the alkoxy O and therefore is a moderate electron donating group via resonance donation of the O lone pair - it's an activating group. The carbonyl in the ketone group is strongly deactivating due to inductive and resonance effects. The halogen group (bromine) is a slightly deactivating group due to inductive effects (due to electronegativity). So the reactivity:
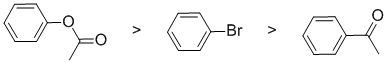
Qu3:
The reactivity of carbonyl groups towards hydride reduction (also relates directly to one of the laboratory experiments). The three functional groups aldehyde, ketone and ester. Since one substituent on each of the carbonyls is the same (the benzene ring), the key factor is how each of the other substituents on each of the carbonyl groups affects the electrophilicity of the carbonyl carbon. In an aldehyde, the group is an H and so is our reference point (i.e. electronically neutral). In the ketone, it's an alkyl group (-R) which is a weak electron donor. In the ester, the alkoxy (-OR) group is a strong electron donor. Electron donors on the carbonyl make the carbon less electrophilic and less reactive. Hence in terms of reactivity:
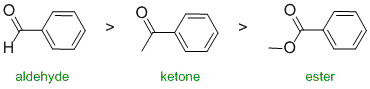
Qu4:
Resonance energy indicates the stability of the conjugated system. Aromatic systems have high resonance stablisation and therefore high resonance energies. Conjugated systems also have resonance stabilisation but not to the same extent as aromatics. Benzene perhaps our "model" aromatic compound (resonance energy = 36 kcal/mol). Styrene (phenylethene) has a benzene ring in conjugation with an alkene and therefore it will have more resonance energy than benzene itself. Cyclopentadiene which is not aromatic, is just a conjugated diene (4 kcal/mol) . Hence:
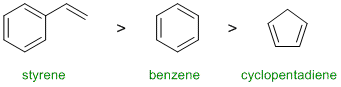
Qu5:
First identify the alpha positions, the positions adjacent to the carbonyl groups. C-H systems in the alpha positions are enolisable H. In the diagram below, the C-H in the alpha positions are shown in blue:
Qu6:
The systems are substituted alkenes so we are looking at electrophilic addition to an alkene . A closer look at the structures shows that the three systems have similar base structures (i.e. alkenes) each with a different substituent, an alkoxy group -OR, an alkyl group -R and a nitro group -NO2. Each alkene will react with the acid to give a carbocation and the substituent will affect the availability of the pi electrons: electron donating substituents will make them more available and electron withdrawing will make them less available. Or we can look at carbocation stability.... an electron donor will stabilise a carbocation, an electron withdrawer will destabilise a carbocation. Looking at the substituents: an alkoxy group, -OR, is a strong electron donor, an alkyl group, -R, is a weak electron donor and a -NO2 group is a strong electron withdrawing.
![]()
Qu7:
Oxidation states...The easiest way to reach the answer is based on the knowledge that nitriles can be viewed as carboxylic acid derivatives (same oxidation state) and that carboxylic acids can be reduced to aldehdyes which in turn can be reduced to alcohols.
More formally, we count the bonds attached to the atom being considered. A bond to a more electronegative atoms counts -1, a bond to the same type of atom counts 0 and a bond to a less electronegative atom (e.g. H) counts +1. Total the count and then consider the formal charge on the central atom since the oxidation state for the central atom plus the groups attached must equal the atoms formal charge. In the CN the C is attached to 3 x N (count - 1 each, total -3) and C (count 0) therefore total = -3 and therefore the oxidation state C = +3. In the C=O the C is attached to 2 x O (count - 1 each, total -2), C (count 0) and H (count +1) therefore total = -1 and therefore the oxidation state C = +1. In the alcohol the C is attached to O (count - 1 each, total -1), C (count 0) and 2xH (count +2) therefore total = +1 and therefore the oxidation state C = -1.
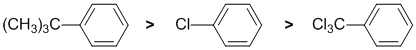
Qu8:
The reaction is an example of electrophilic aromatic substitution, a bromination. We need to look at the substituent effects on the aromatic ring. The substituents are a t-butyl group (large alkyl group), -Cl and a trichloromethyl, -CCl3. The t-butyl group (-R) is slightly electron donating groups via hyperconjugation - it's an activating group and ortho/para directing but the tert-butyl group is large - it's steric size will favour para since the ortho sites are partially blocked. The -Cl is weakly deactivating due to inductive effects, but still directs ortho/para. However, the steric size of the t-butyl group will increase the para yield. In contrast, the trichloromethyl group acts as an electron withdrawing group due to the inductive effects caused by the electronegativity of the chlorine atoms, and electron withdrawing groups direct meta.
Qu9:
The reactivity of carboxylic acid derivative carbonyl groups of an acid anhydride, an amide, and an ester towards hydrolysis. The electronic factors of each of the substituents / leaving group on each of the carbonyl groups need to be considered. In the anhydride, OC(+O)R, the carboxyl group, is a moderate electron donor and a reasonable leaving group. In the amide, the NHR group, is a strong electron donating group and a very poor leaving group. In the ester, the -OR group, the methoxy group is a strong electron donor and a poor leaving group. Electron donors on the carbonyl make the carbon less electrophilic and less reactive, a better leaving group makes the acid derivative more reactive. Hence in terms of reactivity:
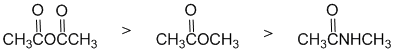
Qu10:
The question is looking at the formation of Grignard reagents, where the reactivity mirrors the length and strength of the C-X bond (the weaker the bond the more readily the Grignard reagent, R-Mg-X forms, so i > Br > Cl.
STRUCTURES and PROPERTIES:
You need to know about functional groups and reactions, and how to apply concepts related to structure such as stereochemistry, acidity/basicity and reactivity etc.
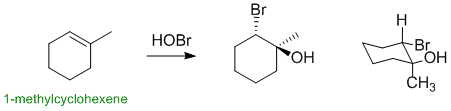
Qu11:
1-methylcyclohexene reacts with HOBr via an electrophilic addition to give a halohydrin. The addition occurs via a cyclic halonium ion where the HO- (Nu) adds to the more substituted positions and with overall anti stereochemistry. This means the HO- needs to be on the same C as the methyl group and the HO- and Br- need to be trans- to each other (opposite sides of the ring):
Qu12:
Epoxides can be formed from trans-1,2-halohydrins (trans allows the correct 180 degree geometry for the SN2 reaction). The NaH (hydride) reacts as a base making a better nucleophile:
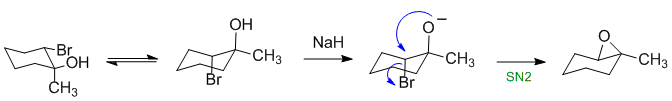
Qu13:
1-methylcyclohexene reacts with aq. sulfuric acid via an electrophilic addition undergoing hydration to give an alcohol. The major product is the Markovnikov alcohol (HO- at the more highly substituted position):
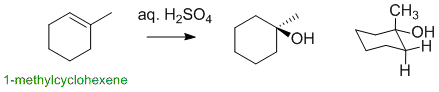
Qu14:
1-methylcyclohexene undergoes hydroboration / oxidation (an electrophilic addition) to give an alcohol. The major product is the anti-Markovnikov alcohol (HO- at the more less substituted position), and the overall addition occurs with syn stereochemistry. This means the HO- and H- need to be cis- to each other (same side of the ring):
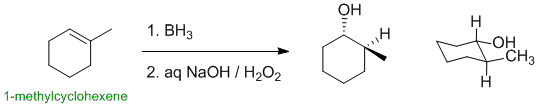
Qu15:
In order for the HO- group to be cis to a methyl group, the two groups need to be on the same face of the ring structure.
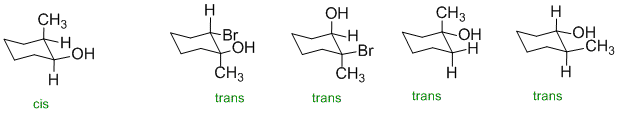
Qu16:
Knowing the pKa's really helps.... The crux here is knowing that enolates that have two electron withdrawing groups are more acidic than alcohols and that ketones are better electron withdrawing groups than esters.
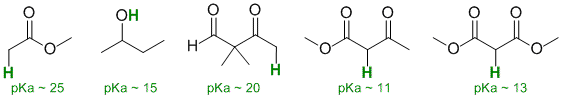
Qu17:
A methylene is a CH2 group (cf a CH3 is a methyl group and CH methine). Active methylenes are CH2 groups between two electron withdrawing groups. They are highlighted in blue in the figure shown:
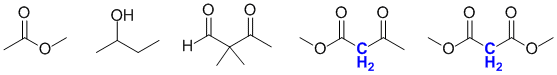
Qu18:
Of the functional groups present in the potential starting materials, ketones, aldehydes and esters all reduce with LiAlH4 to give alcohols and in particular, the C=O in aldehydes and esters will give primary alcohols and ketones secondary alcohols. Draw the product, then count C atoms to be sure.
Qu19:
Counting H types:
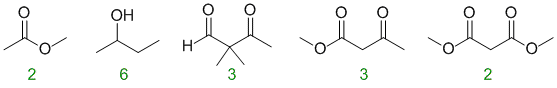
AROMATICITY and RESONANCE:
Best method is to work through the molecules and decide on the aromaticity based on the four criteria for the pi system (cyclic, planar, conjugated pi system with 4n+2 pi electrons).
Qu20:
The Huckel rule requires that there are 4n+2 pi electrons in the cyclic pi system. The questions says n is not 1, so for n=0 this means 2 pi electrons, n=2 means 10 pi electrons....

Qu21:
To be non-aromatic as drawn but with an aromatic resonance structure, we need to find a system that satisfies all the criteria for aromaticity except the electron count and we can therefore add / subtract electrons via resonance. Perhaps most critically, it means that all the atoms in the cyclic structure must already contribute to the p system. So in these examples, it means they are all sp2 hybridised.
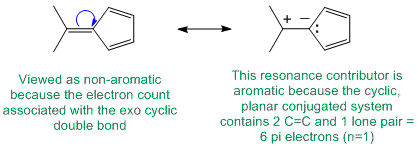
Qu22:
To be non-aromatic as drawn but with an aromatic conjugate base, we need to find a system that becomes aromatic when we remove a proton and form the conjugate base typically adding another pair of electrons to the pi electron count for the Huckel rule.
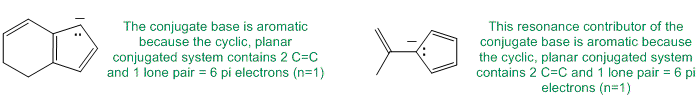
Qu23:
Polyenes have more than one C=C. Resonance stabilisation comes from conjugation. Aromatic systems have higher resonance stabilisation so an aromatic systems with the most C=C will have the most resonance stabilisation.
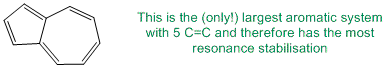
Qu24:
Polyenes have more than one C=C. Resonance stabilisation comes from conjugation, so a non-conjugated i.e. an isolated polyene would have no resonance stabilisation.

Qu25:
The most basic N will have a lone pair that is the most available to donate to a proton. The availability of the lone pair will be reduced (i.e. less basic) if it is part of the aromatic system (i.e. delocalised). The nature of the orbital the lone pair is in also has an impact due to proximity to the +ve nucleus so in terms of availability, sp3 > sp2. Only one of the structures contains an sp3 hydridised N.

Qu26:
To be aromatic as drawn but with a non-aromatic conjugate acid, we need to find a system that is aromatic as drawn yet when it reacts with H+ is no longer aromatic. A typical example would be where a lone pair of a heteratom is initially part of the aromatic pi system but those electrons are used to make the new bond to the H+.
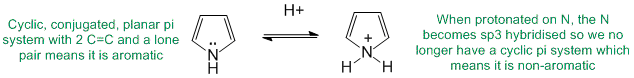
Qu27:
To be non-aromatic as drawn but with an aromatic resonance structure, we need to find a system that satisfies all the criteria for aromaticity except the electron count and we can therefore add / subtract electrons via resonance. Perhaps most critically, it means that all the atoms in the cyclic structure must already contribute to the p system. So in these examples, it means all the atoms are sp2 hybridised.
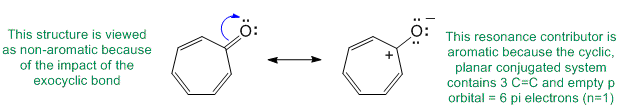
Qu28:
In order to be sp3 hybridised, it needs to be only involved in single bonds and not be interacting through resonance with an adjacent pi system (i.e. not conjugated).
Qu29:
The most common form of tautomerism switches H and double bond positions and the involvement of carbonyl groups is quite common. Tautomers of ketones are the enols.
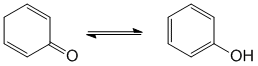
STARTING MATERIALS AND PRODUCTS:
If you are trying to find the product, then you should probably just work forwards through the sequence of reactions.
If you are looking for the starting material, then working backwards is probably the best way to go....
Basically depends on the need to know and identify the reactions, this is often triggered by looking at the functional groups in the molecules.
Qu30:
Working forwards, treatment of a 1,2-halohydrin with sodium carbonate as a base forms the epoxide via an SN2 reaction. This means that we need to set up the starting material in the conformation in which is reacts where the HO and the Br are anti to each other so that the Nu can attack the backside (at 180 degrees to the LG) and cause the normal SN2 inversion. A, B and Call have the wrong functional groups. D has the wrong stereochemistry because you've ignored the SN2 stereochemical requirements.
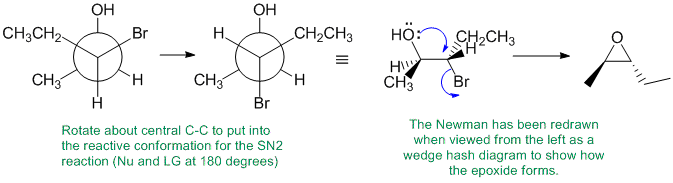
Qu31:
Working forwards, the terminal alkyne undergoes hydration to give a methyl ketone. Hydride reduction of the ketones gives a secondary alcohol.
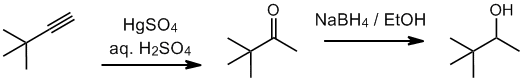
Qu32:
Working forwards, a conjugated diene and trans-(di)substituted alkene (the dienophile) .... it's a Diels-Alder reaction. The hydride reagent in step 2 will reduce the ester groups to primary alcohols (after work up = step 3). Then in step 4, catalytic hydrogenation, reduce the alkenes C=C to an alkane C-C. This is a syn reduction so the product is the primary trans-1,2-diol.
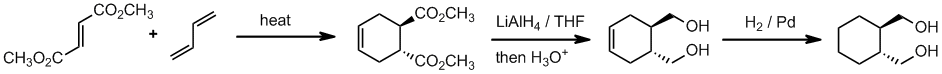
Qu33:
Working backwards, the product has a primary amine and alcohol after reduction. Looking at step 2, looks like cyanide as a nucleophile which is consistent with the reduction to generate the primary amine, -CH2NH2. Since we have an aromatic system, the substitution is of a diazonium salt formed in step 1 from the initial aromatic amine:
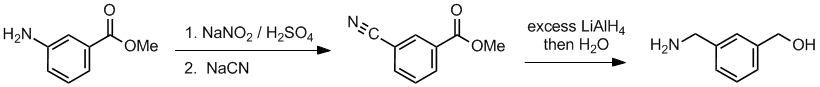
Qu34:
Working forwards, the ketone will undergo a Baeyer-Villager reaction to give the cyclic ester (O inserts on the more substituted side) followed by transesterification (with work up) resulting in ring opening.
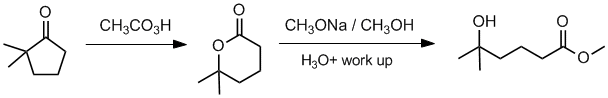
Qu35:
Working forwards, the peracid reacts with the cis-alkene to give an epoxide where the methyl groups are cis. Aq. acid will then open the epoxide to give a 1,2-diol via the equivalent of an anti addition to give a diol with the stereochemistry shown. The 1,2-diol reacts with a ketone to give a cyclic ketal
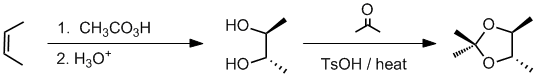
Qu36:
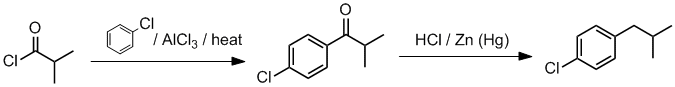
Working backwards, in step 2, we have done a Clemmensen reduction to convert a C=O into a CH2. The first step is an electrophilic
aromatic substitution, specifically Friedel-Crafts
acylation or alkylation (based on the AlCl3) but we see the aromatic unit, so what did we add ? Based on the use of the Clemmensen and the nature of the alkyl group, we would need to acylate to avoid a rearrangement.
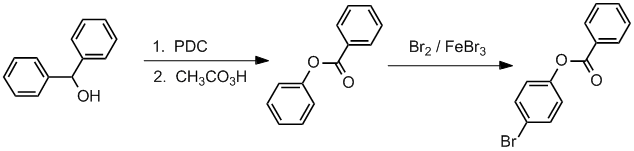
Qu37:
Working forwards, the first step is PDC oxidation of the secondary alcohol to the ketone. The ketone will then undergo a Baeyer-Villager reaction to give the ester. Finally, an electrophilic
aromatic substitution, specifically bromination on the more activated ring mainly in the less hindered para-position.
Qu38:
Working forwards, the first step is the reaction of a Grignard reagent with an epoxide (reacts at the least hindered end) which after work up gives a secondary alcohol... count carbons.
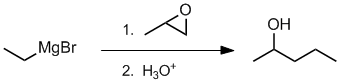
REAGENTS FOR SYNTHESIS:
Need to be able to look at reactions, looking at the functional groups in the starting materials and products of each step to think about
how you have got there.
How well do you know your reagents
? Look at what has actually happened in terms of the reaction functional
group transformation and then first look for any regiochemical issues
then finally the stereochemistry last (it's the hardest to sort out).
Qu39:
Oxidise methanol to methanal using PDC (good for stopping at the aldehyde).
Qu40:
The conversion of methanal to ethanol, requires adding a C atom.... use Grignard CH3MgBr the H+
Qu41:
Oxidise ethanol to the carboxylic acid using aq. acidic dichromate.
Qu42:
Convert the carboxylic acid to the acid chloride with thionyl chloride, SOCl2.
Qu43:
Friedel-Crafts
acylation between an acid chloride and benzene to make the aromatic ketone.
Qu44:
A Baeyer-Villager reaction by a peracid, RCO3H, converts the ketone to the ester.
Qu45:
Aromatic nitration using HNO3 / H2SO4
Qu46:
Reduce the nitro group to an aromatic amine using Sn / HCl then base.
Qu47:
Convert the amine to the amide by reacting with the acetic anhydride, (CH3CO)2O.
Qu48:
Hydrolysis of the ester gives the alcohol but without hydrolysing the amide using aq. base / heat.
EXPLANATION OF PHENOMENA
Qu51:
It's the group on the ring that directs the substitution, deactivators direct meta. The –CO2H group is deactivating and a m-director.
Qu52:
The reactivity of carbonyl groups towards nucleophiles depends on the electrophilicity of the carbonyl carbon which is controlled by the electronic or steric effects of the attached groups. The F atoms in hexafluoropropane make the carbonyl more reactive due to the electronegative effects of the F via the sigma bonds (induction).
Qu53:
The equilibrium between a ketone and ketal is normally acid catalysed. Under basic conditions, a ketal would lack a good leaving group.
Qu54:
The difference between the two amides is that the most acidic H in X is an N-H system, while in Y it is a C-H system. Hence, X is more acidic due to the difference between the N and the C.
Qu55:
Aldehydes are more reactive than ketones because the extra alkyl group in the ketone makes the C=O C less electrophilic due to steric and electronic effects.
![[Chem 353 Home]](../mol.gif) Return to Homepage
Return to Homepage